[This is the second post in a series designed to help you memorize the amino acids without mnemonics. It covers glycine, alanine, serine, cysteine, threonine, and proline, with a recurring theme of stereochemistry.
The first part is here, and it is the introduction. You should read it if you are wondering why there is so much detail here above and beyond simply stating what the amino acids are, and why I think this excess of detail will help you memorize the amino acids. I’ll just say here that it is to help you fit them into a larger system in your mind, and you don’t have to memorize all this detail in order to memorize the amino acids.
The third part of this series will cover phenylalanine, tyrosine, valine, leucine, isoleucine, methionine, and tryptophan. The fourth part will cover histidine, lysine, arginine, aspartate, asparigine, glutamate, and glutamine. After I publish the later parts, I will update the contents of these brackets accordingly.]
Which amino acids are “the amino acids”?
An amino acid is any molecule that has both a carboxylic acid group and an amine group, so there are infinitely many possible amino acids. Here is an example:
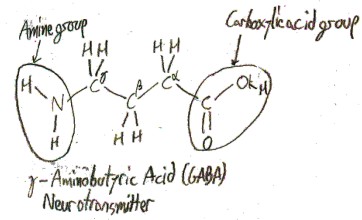
When an amino acid is dissolved in water, as it often is in biology, most of the amine groups become protonated and most of the carboxylic acid groups become deprotonated. It then looks like this most of the time:
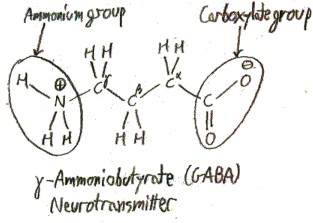
Though it would be technically correct to call the protonated amine group an “ammonium” group and the deprotonated carboxylic acid group a “carboxylate” group, as shown in this picture, this is seldom done. Unless they are specifically talking about protonation states, chemists will talk loosely and refer to the “amine group” or the “carboxylic acid group” of an amino acid, even when it is dissolved in water.
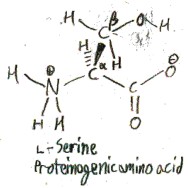
So far, it seems like there must be a lot of amino acids. However, when we talk about memorizing the “amino acids”, we are just talking about the proteinogenic amino acids, which are amino acids that are incorporated into proteins during translation, the process whereby ribosomes read RNA and construct proteins accordingly. All proteinogenic amino acids are α-amino acids, which means that their amine group is located on the α-carbon, which is the carbon bonded to the carboxylic acid group. (The β-carbon is the carbon bonded to the α-carbon, and the γ-carbon comes after that, etc.)
Here are some proteinogenic amino acids connected together in a protein.
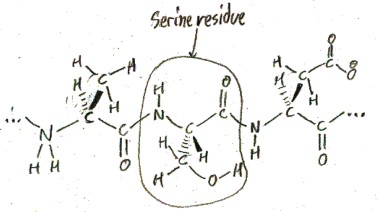
When amino acids are in a protein like this, they are called “amino acid residues”. When they are not in a protein, they are called “free amino acids”.
All proteinogenic amino acids also have an α-hydrogen, a hydrogen bonded to the α-carbon.
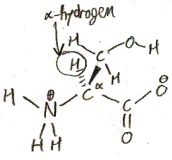
So far we have talked about 3 of the 4 substituents on the α-carbon: the carboxylic acid group, the amino group, and the hydrogen. The fourth substituent is what differs in the different amino acids. In principle, there are infinitely many possibilities for this fourth subsitutuent, but only 20 of these are proteinogenic. So we are going to talk about these 20 in what follows.
Serine
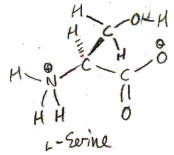
We have already shown the proteinogenic amino acid serine in the pictures above. Its side chain is hydroxymethyl, or -CH2OH. In 1865 E. Cramer first isolated serine from a silk protein called sericin. Cramer named both serine and sericin, and he named them after sericus, the Latin word for “silk”.
The 3-letter abbreviation of serine is “Ser”, and the 1-letter abbreivation is “S”. The purpose of these abbreviations is to make it easier to list long sequences of amino acid residues in proteins.
Serine is the only amino acid to feature a primary alcohol (an -OH group on a primary carbon, which is a carbon bonded to 1 carbon atom and 2 hydrogen atoms). We will get to other amino acids, which feature a secondary alcohol (threonine) and an aryl alcohol (tyrosine). These three are the only hydroxy amino acids. It is remarkable that there is exactly one of each of these different kinds of alcohols, so that a small number of amino acids can cover a wide range of functions.
Serine exists in two forms: l-serine and d-serine:
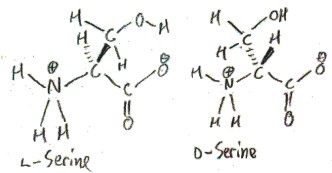
These forms are called enantiomers, from Greek ενάντιος/enantios meaning “opposite” and μέρος/meros meaning “part”. How are they opposite? One of them is the mirror image of the other, just as your left hand is the mirror image of your right hand. The enantiomers of alanine are distinguished by the “l-” and “d-” prefixes. Only l-serine occurs biologically. In fact, all proteinogenic, and most biological, amino acids are l-amino acids. This is an amazing fact! How did things evolve so that life uses only l-amino acids? There is much speculation on this.
To understand what the designations l and d mean will require a digression.
Digression to explain what l and d mean
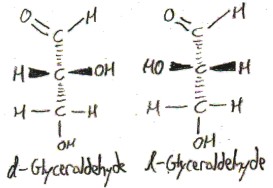
Consider the following sugars:
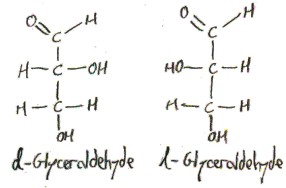
These are drawn in Fischer projection, which means that the vertical lines are viewed as curling backwards into the screen, and the horizontal lines are viewed as curling forwards out of the screen. In order to show this, we draw bonds that are coming out of the screen towards you as wedges and we dash bonds that are going into the screen away from you.
The four substitents of the central carbon atom point, approximately, to the vertices of a tetrahedron centered at it:
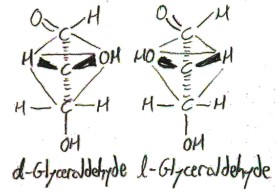
The “d-” and “l-” prefixes distinguish the two enantiomers of glyceraldehyde. I don’t really understand this atm, but they refer to how the compound interacts with plane-polarized light: d-compounds rotate it clockwise (which is called “dextrorotation”, hence “d“) and l-compounds rotate it counterclockwise (which is called “levorotation”, hence “l“).
The problem with the d/l nomenclature is that at the time that it was invented, there was no way to figure out which of the two possible structures corresponded to d and which corresponded to l.
Due to this inadequacy, the German chemist Emil Fischer started to use the d and l designations in a different way. He was dealing with sugars and the molecules that could be synthesized from them, and in this area the inadequacy mentioned really mattered. First he guessed (!) that the structure of d– and l-glyceraldehyde were as shown, as opposed to the other way around. There was a 50% percent chance that he would be right. (In 1951 it was determined by X-rays that he was right!) Once he had made this guess, Fischer began to call molecules that were synthetically related to d-sugars, such as d-glyceraldehyde, d themselves, and similarly for l, regardless of how they interacted with plane-polarized light.
In 1905, the Russian-American chemist Martin André Rosanoff realized the ambiguity of these two opposing usages of d and l, and proposed to separate them. He suggested that d and l be restricted to their original usage, relating to plane-polarized light, and that the Greek letters δ and λ be used instead for Fischer’s new usage, to refer to molecules that were synthetically related to d– and l-sugars. He also resolved another ambiguity: sometimes the same molecule could be synthesized from both an l-sugar and a d-sugar, so that it wasn’t clear whether to call it δ or λ. He resolved this ambiguity by restricting the possible syntheses that were allowed.
What we do today is a further variation of Rosanoff’s contribution. First of all, instead of using the Greek letters δ and λ, we use the small caps d and l. Secondly, instead of referring to a synthetic relationship between the molecule in question and d– or l-glyceraldehyde, d and l refer to a resemblance in the structure, regardless of how the molecule was synthesized. This is called the Fischer-Rosanoff convention, after Fischer and Rosanoff.
Here is how this works in the case of serine. We draw serine in Fischer projection, so that it looks like glyceraldehyde. The enantiomer that “looks like” d-glyceraldehyde is called d, and the one that “looks like” l-glyceraldehyde is called l.
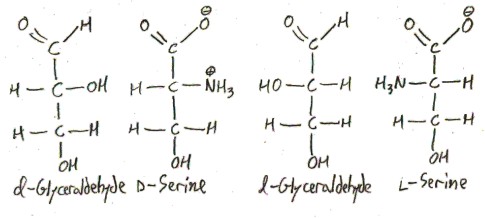
For more information on this (I think, fascinating) subject, read the articles I read to write this section!
- D. W. Slocum, D. Sugarman, and S. P. Tucker 1971: “The Two Faces of d and l Nomenclature”, Journal of Chemical Education, Volume 48, Number 9, pp. 579-600.
- This is a survey article, which cited the following one. The Journal of Chemical Education is great! It is one of the few places that chemists actually generalize and say why they care about the things they care about.
- M. A. Rosanoff 1906: “On Fischer’s Classification of Stereo-Isomers”, Journal of the American Chemical Society, Volume 28, pp. 114-121.
- This is Rosanoff’s original article, where he proposed the nomenclature reform.
- C. S. Hudson 1941: “Emil Fischer’s Discovery of the Configuration of Glucose”, Journal of Chemical Education, Volume 18, Number 8, pp.353-357.
- Not directly relevant, but an exposition of the absolutely beautiful work Fischer was doing, and why it so heavily involved stereochemistry.
Note: Just as life primarily uses l-, rather than d-amino acids, it also primarily uses d-, rather than l-sugars.
Alanine
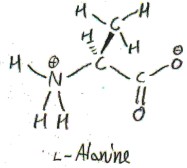
If we replace the hydroxyl group in serine by a hydrogen atom, we obtain alanine. Its side chain is just a methyl group (methyl is -CH3).
To see why l-Alanine is called l, we show it next to l-glyceraldehyde:
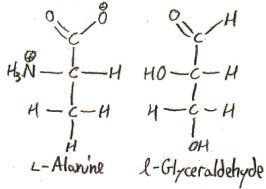
It doesn’t line up as perfectly as serine, because there is no hydroxyl anymore, but we still line it up the same way, because that’s the Fischer-Rosanoff convention.
It was first synthesized, not isolated. The German chemist Adolph Strecker synthesized it from acetaldehyde in 1850, which in his time (and in German) called simply “Aldehyd”. Here was his synthesis, along with his names for the compounds involved and their modern names:
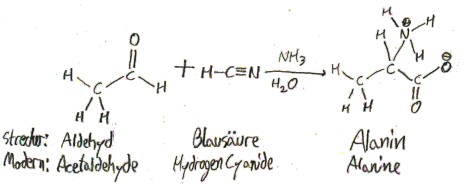
Strecker named alanine “Alanin” because it was derived from “Aldehyd”.
The synthesis of alanine from acetaldehyde in fact generalizes to the synthesis of any amino acid from the corresponding aldehyde, and this synthesis is named (aptly) the Strecker synthesis. For example, here is how Fischer, in 1902, synthesized serine from the corresponding aldehyde:
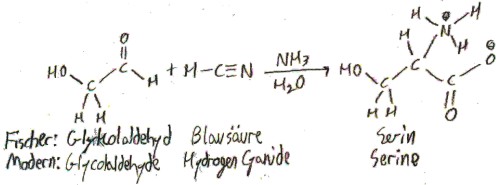
Alanine is the most “normal” amino acid. Thus it often functions as a “control” in experiments on proteins, in the sense that biochemists will mutate a residue of a protein to alanine to see what happens when the specific function of that residue is absent.
The 3-letter and 1-letter abbreviations are just what you would think: “Ala” and “A”.
Glycine
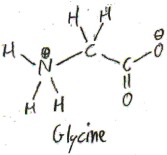
Glycine is the simplest amino acid. Its side chain is merely a hydrogen atom.
Glycine is achiral, which means that it is superimposable over its mirror image. To see this, we can draw the following Fischer projection of it:
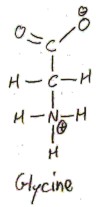
If we reflect this about a vertical mirror, we get the same thing! Thus l-glycine and d-glycine are actually the same, so we just say “glycine”, without any prefix. Due to its achirality, glycine does not rotate plane-polarized light either clockwise or counter-clockwise, so it is called optically inactive.
On account of its minimal side chain, glycine can rotate more freely than other amino acids, so a protein has enhanced flexibility at glycine residues. This is why it is alanine, and not glycine, that functions as a “control”, as I mentioned in the previous section.
Glycine was isolated from gelatin in 1820. It was named after the Greek word γλυκύς/glykýs, meaning “sweet-tasting”, because of its sweet taste. This is the same root as “glucose”, “glycogen”, and many other words having to do with sweetness.
The 3-letter and 1-letter abbreviations, again, are just what you would think: “Gly” and “G”.
Cysteine
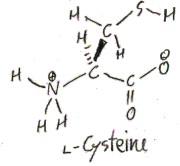
Cysteine is like serine except instead of oxygen it uses the congener sulfur (a congener of an element is an element in the same column of the periodic table). Its side chain is a primary thiol, CH2SH. (A thiol is like an alcohol but with sulfur instead of oxygen. In general, the “thio” prefix denotes that an oxygen is replaced with a sulfur. It comes from the Greek word for sulfur, θεῖον/theîon.)
It was first described in 1810 by Wollaston, and given its name by Berzelius. Its name comes from “cyst”, which is an old term for the bladder, since cysteine was first isolated from urine.
Cysteine also exists in a dimerized form, as cystine. Cystine is really two amino acids, which have a disulfide side chain -CH2S-SCH2– connecting them. Here is how it would appear as a residue in a protein.
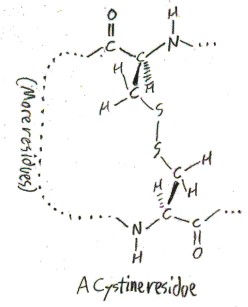
It is not clear what the difference in pronunciation is between “cysteine” and “cystine”. I have heard chemists pronounce “cysteine” simply as /sɪs.tin/ (sis-teen), but I have also heard them make a big point of the distinction by saying /sɪs.tə.ʔin/ (sis-tuh-een).
Cystine adds another level of structure to proteins, because it can cross-link two different sections of the protein together. This is how hair keeps its shape, as straight or curly or whatever. Perms work by chemically breaking the disulfide bonds and reforming them in new places corresponding to a different hairstyle.
Here is a general schematic of disulfide bonds breaking and reforming in new places (Cysteine residues/free amino acids are indicated by “Cys”):
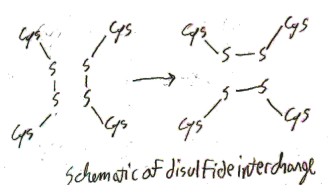
When disulfide bonds are not forcibly broken as in a perm, they generally do not interchange. But when free cystine is exposed to acid, the disulfide bonds do tend to interchange. Surprisingly, this interchange causes transitions between l– and d-cystine! Thus the longer free l-cystine is exposed to acid, the more it will approach a 1:1 mixture of l– and d-cystine, which is called a “racemic mixture”. This interconversion between l– and d-cystine is called racemization, and cystine is the only proteinogenic amino acid that racemizes under acidic conditions. Cystine racemizes because in the process of disulfide interchange the α-hydrogen atom sometimes leaves the molecule briefly and is replace by another hydrogen atom on the other side, like so:
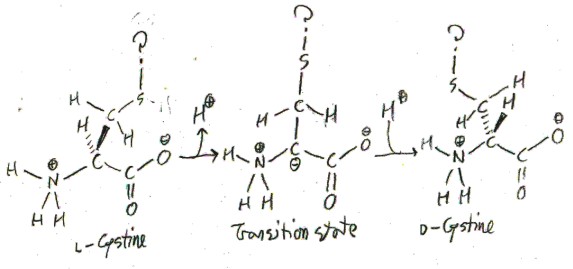
The question marks indicate that this is taking place at sometime in the middle of disulfide interchange, so I’m not exactly sure what the sulfur is bound to. For most amino acids this wouldn’t happen because the α-hydrogen wouldn’t ever leave, but something about the process of disulfide interchange makes this more feasible. One theory is that at some point in the disulfide interchange the sulfur is very electron-deficient, which stabilizes the negative charge on the α-carbon in the transition state.
3-letter and 1-letter abbreviations: “Cys” and “C”. This might seem too obvious to need to state. However, later on the abbreviations aren’t as simple. For example, the 1-letter abbreviation of glutamate can’t be “G” because “G” is already taken by glycine, so it is “Q” instead, because of the pun “Q-tamate”.
Note: If the next congener of oxygen after sulfur, namely selenium, is used, we get selenocysteine, a rare amino acid. It is debated whether selenocysteine is proteinogenic or not, because, although it is coded by RNA, there are some differences between the way it is coded and the way the standard 20 amino acids are coded. For example, the nucelotides that code for it also form the “stop codon”, the sequence that tells ribosomes to stop adding new residues to a protein, and release it. The abbreviations for it are “Sec” and “U” (the latter was apparently chosen because it was the only letter left).
Threonine
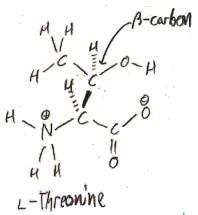
Threonine, as was briefly mentioned above, is a secondary alcohol. This means that it has a hydroxy group on a carbon that is bonded to two other carbons and a hydrogen. The side chain of threonine is serine with a methyl added to the β-carbon, so it really is the minimal way to create a secondary alcohol amino acid.
You may realize that the β carbon has 4 different substituents, so that there are two non-equivalent possible configurations of l-threonine.
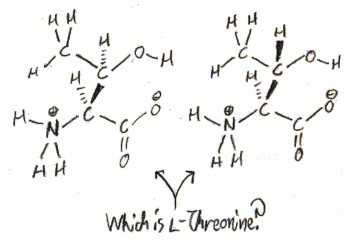
A carbon that can have two non-equivalent configurations, like this, is called a “chiral carbon”, from Greek χείρ/kheír, meaning “hand”, as in right- and left-handed. Thus, threonine is the first amino acid we have discussed to have two chiral carbon (the only other such proteinogenic amino acid is isoleucine). The two configurations of l-threonine are not enantiomers, because the enantiomer of l-threonine would be d, rather than l. We call these two configurations of l-threonine “diastereomers”, rather than “enantiomers”, for this reason.
Only one of threonine’s two diastereomers appears in nature, and its name, “threonine”, as we will see, tells us which one!
Remember how the “l” designation referred to a comparison with “l-glyceraldehyde”? Well, an old form of stereochemical nomenclature extends this comparison, so that diastereomers are named after larger sugars whose structures they are comparable to.
Consider the 4-carbon aldoses, that is, the sugars of the form CHO(CHOH)2CH2OH. There are four such sugars: d-erythrose, l-erythose, d-threose, and l-threose. Here they are shown with d– and l-glyceraldehyde, from which they get their d and l designations by the Fischer-Rosanoff convention:
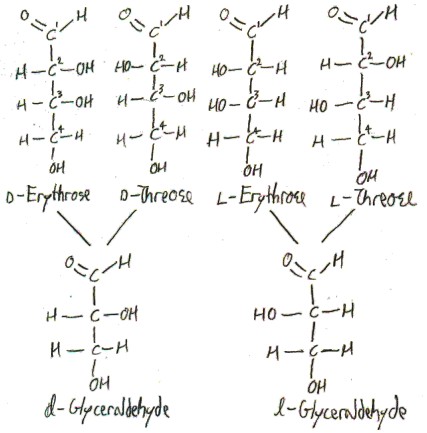
The ” d” and “l” refer to whether the 3-carbon’s configuration is like d– or l-glyceraldehyde, and the rest of the name describes the 2-carbon’s configuration. In erythrose, both of these carbons have the same configuration (in a Fischer projection). In threose, they have opposite configurations. The name “erythrose” comes from the Greek word ἐρυθρός/erythrós for “red”, because it is red in the presence of alkali metals. In a common trick of chemical nomenclature, the diastereomer threose is named by switching the letters of “erythrose”.
Finally, let’s get back to threonine. We can draw the proteinogenic diastereomer in a “Fischer projection” to look kind of like d-threose. This is why it is called “threonine”, rather than “erythronine”. (Here is the paper where they figure out that it is threo- instead of erythro-.)
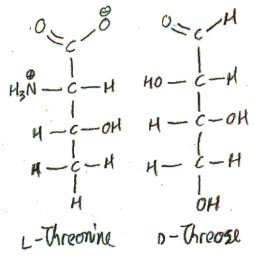
Confusingly, we have drawn an l-amino acid to look like a d-sugar! This is possible because the α-carbon of l-threonine now corresponds to the 2-carbon of the sugar, which is not the carbon of the sugar that makes it a d-sugar. Here we draw l-threonine and d-threose along with their corresponding glyceraldehydes:
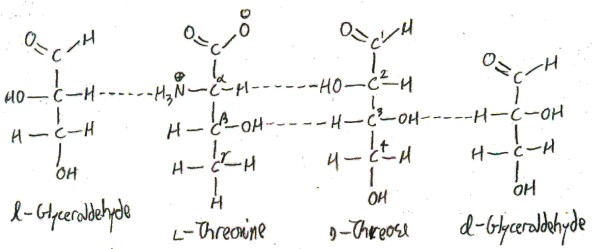
Notice that it is the 3-carbon of d-threose that makes it d, but the α-carbon of l-threonine that makes it l. These carbons don’t correspond to each other in our correspondence, so there is no contradiction.
IUPAC noticed this confusion, so they recommended subscripts on “l” and “d” symbols to disambiguate situations like this. With this system, threonine is ls, but dg. The subscripted “s” stands for serine, and it indicates that threonine is l in the usual system of amino acid nomenclature. This is because, as we noted above, serine is the amino acid that most naturally can be drawn to look like glyceraldehyde. The subscripted g stands for glyceraldehyde, a sugar, and it indicates that the sugar threonine most closely corresponds to is d.
“Erythronine” never became an accepted trivial name, because it is very rare biologically, so the name is generally used instead is “allothreonine”, from Greek ἄλλος/állos, meaning “other”. In general, the prefix “allo-” picks out whatever the other diastereomer than the standard one is.
Threonine is the first amino acid we have discussed that has C-beta branching, which is when the β-carbon is bonded to more than two atoms that aren’t hydrogen. C-beta branching makes it harder for residues to form α-helices.
3-letter and 1-letter abbreviations: “Thr” and “T”.
Proline
Here is proline:
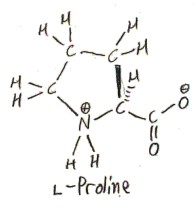
It is often said that proline is not technically an amino acid, but rather an imino acid. This is because the side chain connects to not only the α-carbon, but to the nitrogen atom as well, so that the nitrogen is bonded to two carbons. However, according to IUPAC, the usage of “imino acid” for this situation is obsolescent (declining). The general term “imine” (not specifically about carboxylic acids) refers to a C=N double bond, and the usage of the term for nitrogen bonded to two different carbons is obsolete (not just obsolescent). So I guess the obsolete usage is tolerated when applied to carboxylic acids, because people like to call proline an imino acid!
The side chain of proline consists of three methylenes (methylene is -CH2-), the last of which joins to the nitrogen. This makes a 5-membered ring: Cα, N, and the 3 methylenes. If this ring stood alone, as (CH2)4NH, it would be called pyrrolidine.
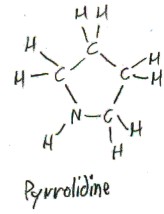
“Proline” is a contraction of “pyrrolidine”. “Pyrrolidine” itself comes from “pyrrole”, which is the aromatic compound (CH)4NH.
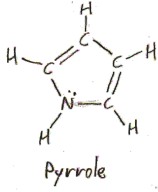
The name “pyrrole” comes from the Greek πυρρός/pyrrhós, meaning “red”, because of the “red color developing when pyrrole vapor acts on a pine splinter moistened with hydrochloric acid” (Elsevier’s Dictionary of Chemoetymology), and the “idine” suffix in “pyrrolidine” denotes saturation (adding the most hydrogen atoms possible while mantaining the structure otherwise). We previously encountered another Greek word meaning “red”: ἐρυθρός/erythrós, which “erythrose” comes from. The difference between these two “reds” is that πυρρός/pyrrhós is a “fiery red”: it comes from πυρ/pyr, meaning “fire”, which the root of English “pyro-“.
The ring in proline severely restricts the rotation of the Cα-N bond. This is an effect that can be considered opposite to that of glycine, whose trivial side chain allows for much more rotation. Proline thus has a unique structural function. It is used for making quick turns, I think. Many statements about amino acid residues must be qualified by “except proline”. For example, there are many proteases which cut proteins at sites where there are specific residues, except if proline is there!
Why does proline have a 5-membered ring, as opposed to some other size? A general rule-of-thumb is that 5- and 6-membered rings are the most stable, so when given the choice, rings tend to have 5 or 6 members. The homolog of proline with a 6-membered ring is called pipecolic acid, and it does appear in biology, but not in proteins, so it is not a proteinogenic amino acid. One possible explanation for nature selecting proline over pipecolic acid is that proline is more rigid since its ring is smaller, so it better executes its unique structural function.
3-letter and 1-letter abbreviations: “Pro” and “P”.





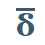
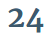
 and
and  are distinct points on a line in the plane, then
are distinct points on a line in the plane, then 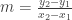 is the slope of the line.
is the slope of the line.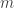 is the slope of a line in the plane and
is the slope of a line in the plane and 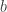 is the y-intercept (i.e. the line intersects the
is the y-intercept (i.e. the line intersects the 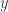 -axis at
-axis at  is an equation for the line.
is an equation for the line. is a point on a line in the plane, then
is a point on a line in the plane, then 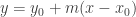 is an equation for the line.
is an equation for the line.

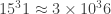 (if you don’t see how I got this, comment and I’ll explain). So if we think of a simple rule, the odds that that rule will happen to generate this random sequence are
(if you don’t see how I got this, comment and I’ll explain). So if we think of a simple rule, the odds that that rule will happen to generate this random sequence are  in
in  . This is extremely unlikely.
. This is extremely unlikely.